
责编 | 兮
在基础物理课上学生经常遇到的一个问题是,给定一个物体受力情况有关参数,比如质量,重力加速度等,求解物体的运动轨迹。学生需要选定一组坐标(动力学变量)表征物体状态(位置 x,数学上可用一个向量表示),利用牛顿力学定律列出坐标的动力学方程。给定初始条件,这组动力学方程就能被用来预测物体任意时刻的坐标。这是动力系统理论的一个简单应用,而实际上大到天体,小到微观粒子,都可以用动力学方程来描述它们的状态随时间的演化。
细胞也是个动力学系统。细胞高通量测序技术使得研究者能够用全基因组表达水平来表征细胞的内部状态 (x),但是测序时因为细胞需要被固定,导致我们不能够连续追踪同一个细胞。如何从固定细胞的基因组数据提取动力学信息成了一个活跃的研究方向。一个重大发展是2018年提出的RNA速度方法。在基因的表达过程中,首先产生的是未剪接的RNA(u),然后其经过剪接形成成熟的RNA(s),最后成熟的RNA再被降解。从测序的结果里我们可以读出每个细胞里未剪接的和成熟的RNA数量,再引入一些假设来估算降解的速率常数(γ),从而进一步计算这个细胞里的每个基因在细胞固定瞬间的RNA速度(ds/dt = u-γs),即这个基因表达是在上调(正速度代表活化),还是在下调(负速度代表抑制)。RNA速度的工作引发了一系列的应用以及方法上进一步的发展。那么能否从这些离散的单细胞瞬时数据重构连续的细胞基因表达调控的动力学方程呢(dx/dt = F(x))?这不仅是理论概念上的突破,也是技术上的一个具挑战性的逆向问题(inverse problem)。
2022年2月1日,由麻省理工学院Jonathan Weissman课题组和匹兹堡大学医学院的邢建华课题组以及其他合作者的联合团队(博士后邱肖杰和博士研究生张衍是共同第一作者,邱肖杰博士后为共同通讯)在Cell上发表了文章Mapping transcriptomic vector fields of single cells,提出并开发了基于机器学习的以动力学系统理论为基础的一整套算法及相应的软件包(dynamo: https://github.com/aristoteleo/dynamo-release)。

动画:矢量场可用于预测人体造血等动力学过程中单个细胞的长期轨迹
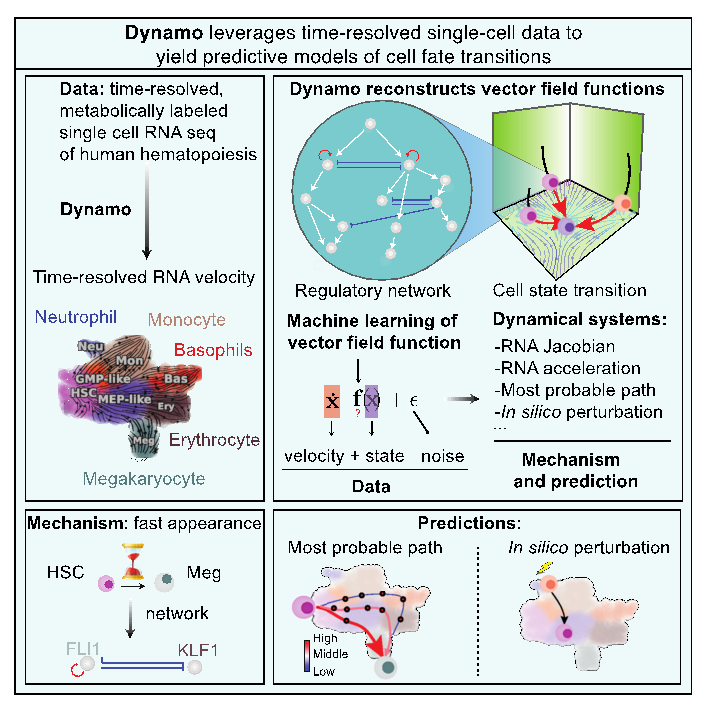
这项工作主要由四个部分组成(图 1)。第一步是从基因表达的生物物理模型出发,建立一套普适的用各种形式的单细胞测序数据估算RNA速度的方法 (图1 上左)。尤其需要指出的是,原始的基于剪接的RNA速度方法只是定性或半定量的给出了估算的相对值,并且对适用体系有很大限制。邱、张等引入了一套新的结合RNA代谢标记的单细胞基因组测序技术的RNA速度定义及计算方法。RNA代谢标记采用化学物质处理活细胞来化学修饰RNA,在测序过程中,诱发其发生碱基T到C的替换。这样可以测得化学处理后新生成的RNA的数量,从而通过化学动力学模型来获得RNA生成或者降解的速率常数,并进一步计算总RNA的速度。邱、张等将两种方法应用到一组人血干细胞分化数据上,结果显示,如果采用传统的基于剪接的RNA速度方法,得到的RNA速度矢量不仅噪音大,还给出了生物上错误的方向(比如从MEP样细胞到巨核细胞的矢量簇),而基于代谢标记的RNA速度则能正确地指向干细胞分化的方向 (图1左上)。
第二步是从离散的单细胞数据重构解析形式的连续的高维动力学方程,dx/dt=F(x) (图1 右上)。函数F(x) 定义了一个向量场,但具体的函数形式很难从数据或生物文献中直接获得。数学上所有可能的相关函数组成了一个无穷维的函数空间(希尔伯特空间)。重构过程就是用机器学习的算法在函数空间中把能最大程度拟合数据的函数给挑出来。这里技术上的一个挑战是高维大数据带来的计算困难。邱、张等采用了在图像处理领域发展的Sparse vector field consensus算法,避免了高维空间的复杂模型需要极大量数据来避免过拟合的问题。
第三步是从学得的向量场提取定量的生物机理信息(图1 左下)。细胞转录向量场就类似于分子动力学模拟中的力场。如果单细胞数据足够精确完备,得到的向量场则包含基因之间相互调控的定量信息。邱、张等通过引入一系列微分几何量(雅可比矩阵、加速度、曲率、散度和旋度等)来提取向量场中隐含的生物信息。造血过程的一个有趣的现象是巨核细胞相较其他成熟细胞类型更早出现。然而,这种现象背后的确切机制仍然是一个谜。在这项研究中,邱、张等试图对该过程提供生物机理上和预测性的理解。他们首先使用基于向量场的伪时间(pseudotime)确认了巨核细胞谱系的早期出现。接下来,他们发现巨核细胞谱系主调节因子FLI1在造血干细胞状态下就已经表现出高表达。此外,雅可比矩阵(Jacobian)分析揭示了FLI1的自我激活,以及FLI1和KLF1(红细胞谱系主调节因子)之间的相互抑制。这些分析共同表明FLI1的自我激活在造血干细胞状态下能够保持其较高的表达,使得造血干细胞首先以高速并加速向巨核细胞谱系产生,同时通过抑制KLF1抑制对红细胞的产生。此外, 在造血干细胞分化的过程中有两个重要的转录因子:GATA1和SPI1(PU.1)。几十年的研究揭示它们相互拮抗又各自自我激活,而这些调控关系都包含在由测序数据重构的向量场中。对向量场的分析给出SPI1对GATA1转录的有效抑制需要达到一定的阈值(希尔系数≈2),但GATA1对SPI1的抑制却接近线性且无阈值(希尔系数≈1)。邱、张等发现的这个GATA1-SPI1非对称的调控机制吻合之前报道的定量实验结果。从一组高质量的单细胞测序数据就能得出过去需要通过多年耗费大量人力物力的实验才能得出的全基因组水平的定量基因调控信息,十分振奋人心。。
第四步是根据重构的动力学方程做预测指导进一步的实验研究(图1右下)。控制细胞形态转化有很重要的医学价值。在2012年中山申弥就因为细胞重组的开创性研究获得了诺贝尔奖。但是现在仍然主要是靠耗时费力的试错法来寻找合适的重组因子。邱、张等展示了可以通过寻找及分析向量场中最小作用量路径来准确预测血细胞不同形态之间转化的重组因子。具解析形式的向量场允许在计算机上预测每个细胞中每个基因的表达响应和分子生物学扰动后的细胞命运变化。有趣的是,抑制GMP谱系的主调节因子SPI1,能使细胞转向巨核细胞和红细胞,而抑制MEP谱系的主调节因子GATA1,能使细胞转向单核细胞和中性粒细胞。然而,抑制这两个基因会使细胞限制在祖细胞状态。这些预测与 (Rekhtman et al., 1999) 中的报道非常吻合,并揭示了SPI1和GATA1 在驱动GMP和MEP谱系中的跷跷板效应调节。
作为总结,这项工作突破了传统的生物信息学方法,对在动力系统理论的框架下分析单细胞生物大数据做了有益的尝试。随着单细胞组学技术测量精度的进一步提高及更多理论及计算工具的应用,在这个框架下重建动力学方程将有很大的发展空间。比如,通过结合固定细胞和活细胞成像的方法(详见BioArt报道:Sci Adv | 邢建华团队开发活细胞追踪图像及定量分析的方法),研究者有望建立更全面、准确的细胞内部状态演化的动力学及机理模型。
原文链接:
https://doi.org/10.1016/j.cell.2021.12.045
转载须知
【非原创文章】本文著作权归文章作者所有,欢迎个人转发分享,未经允许禁止转载,作者拥有所有法定权利,违者必究。


文章评论